Applications
The BAM National Model results are meant to be used in various applications. We provide some R scripts here to facilitate the use of the results.
Prerequisites
We will assume that you use R version >= 3.6 with the following packages installed: raster, sf, jsonlite, readxl, ggplot2, and optionally googledrive.
Working with the JSON API
The JSON API uses JSON as data exchange format. Let’s define the URL for the BAM v4 API:
library(jsonlite)
api_root <- "https://borealbirds.github.io/api/v4"
Get the list of species
First, we need to get the list of species from the JSON API, so that we know what species codes to use:
tab <- fromJSON(file.path(api_root, "species"))
str(tab)
## 'data.frame': 143 obs. of 8 variables:
## $ id : chr "ALFL" "AMCR" "AMGO" "AMPI" ...
## $ idnext : chr "AMCR" "AMGO" "AMPI" "AMRE" ...
## $ idprevious: chr "YTVI" "ALFL" "AMCR" "AMGO" ...
## $ scientific: chr "Empidonax alnorum" "Corvus brachyrhynchos" "Spinus tristis" "Anthus rubescens" ...
## $ english : chr "Alder Flycatcher" "American Crow" "American Goldfinch" "American Pipit" ...
## $ french : chr "Moucherolle des aulnes" "Corneille d'Amérique" "Chardonneret jaune" "Pipit d'Amérique" ...
## $ family : chr "Tyrannidae" "Corvidae" "Fringillidae" "Motacillidae" ...
## $ show : logi TRUE TRUE TRUE TRUE TRUE TRUE ...
head(tab[,c("id", "english")])
## id english
## 1 ALFL Alder Flycatcher
## 2 AMCR American Crow
## 3 AMGO American Goldfinch
## 4 AMPI American Pipit
## 5 AMRE American Redstart
## 6 AMRO American Robin
Get estimates for a species
We can use the id column if we need to loop over multiple species. Now
we’ll only use one species:
spp <- "CAWA"
results <- fromJSON(file.path(api_root, "species", spp))
results$species$english
## [1] "Canada Warbler"
str(results, max.level=2)
## List of 3
## $ species :List of 8
## ..$ id : chr "CAWA"
## ..$ idnext : chr "CCSP"
## ..$ idprevious: chr "BWWA"
## ..$ scientific: chr "Cardellina canadensis"
## ..$ english : chr "Canada Warbler"
## ..$ french : chr "Paruline du Canada"
## ..$ family : chr "Parulidae"
## ..$ show : logi TRUE
## $ popsize :'data.frame': 20 obs. of 4 variables:
## ..$ region : chr [1:20] "Canada" "4 Northwestern Interior Forest" "5 Northern Pacific Rainforest" "6 Boreal Taiga Plains" ...
## ..$ abundance:'data.frame': 20 obs. of 3 variables:
## ..$ density :'data.frame': 20 obs. of 3 variables:
## ..$ areakmsq : num [1:20] 6.21 0.546 0.136 1.18 1.49 1.32 0.0552 0.372 0.444 0.373 ...
## $ densplot:'data.frame': 20 obs. of 2 variables:
## ..$ region: chr [1:20] "Canada" "4" "5" "6" ...
## ..$ data :'data.frame': 20 obs. of 4 variables:
The species element in the list contain the species info saw already
in the species table.
The popsize element contains population sizes, densities, and areas
for the various regions:
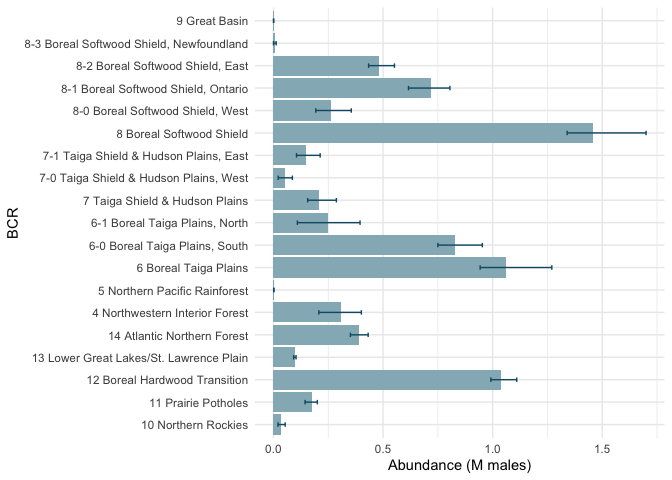
(N <- do.call(cbind, results$popsize))
## region abundance.estimate abundance.lower
## 1 Canada 4.8100 4.5900
## 2 4 Northwestern Interior Forest 0.3090 0.2070
## 3 5 Northern Pacific Rainforest 0.0014 0.0009
## 4 6 Boreal Taiga Plains 1.0600 0.9430
## 5 7 Taiga Shield & Hudson Plains 0.2060 0.1560
## 6 8 Boreal Softwood Shield 1.4600 1.3400
## 7 9 Great Basin 0.0002 0.0000
## 8 10 Northern Rockies 0.0353 0.0208
## 9 11 Prairie Potholes 0.1750 0.1440
## 10 12 Boreal Hardwood Transition 1.0400 0.9920
## 11 13 Lower Great Lakes/St. Lawrence Plain 0.0986 0.0919
## 12 14 Atlantic Northern Forest 0.3880 0.3510
## 13 6-0 Boreal Taiga Plains, South 0.8290 0.7500
## 14 6-1 Boreal Taiga Plains, North 0.2480 0.1090
## 15 7-0 Taiga Shield & Hudson Plains, West 0.0539 0.0217
## 16 7-1 Taiga Shield & Hudson Plains, East 0.1500 0.1050
## 17 8-0 Boreal Softwood Shield, West 0.2640 0.1930
## 18 8-1 Boreal Softwood Shield, Ontario 0.7170 0.6160
## 19 8-2 Boreal Softwood Shield, East 0.4800 0.4340
## 20 8-3 Boreal Softwood Shield, Newfoundland 0.0072 0.0001
## abundance.upper density.estimate density.lower density.upper areakmsq
## 1 5.2100 0.0077 0.0074 0.0084 6.2100
## 2 0.4010 0.0057 0.0038 0.0073 0.5460
## 3 0.0023 0.0001 0.0001 0.0002 0.1360
## 4 1.2700 0.0090 0.0080 0.0107 1.1800
## 5 0.2870 0.0014 0.0010 0.0019 1.4900
## 6 1.7000 0.0111 0.0102 0.0129 1.3200
## 7 0.0011 0.0000 0.0000 0.0002 0.0552
## 8 0.0534 0.0009 0.0006 0.0014 0.3720
## 9 0.2000 0.0040 0.0032 0.0045 0.4440
## 10 1.1100 0.0280 0.0266 0.0297 0.3730
## 11 0.1030 0.0097 0.0090 0.0101 0.1020
## 12 0.4320 0.0199 0.0180 0.0222 0.1950
## 13 0.9530 0.0110 0.0099 0.0126 0.7560
## 14 0.3950 0.0058 0.0025 0.0092 0.4280
## 15 0.0862 0.0010 0.0004 0.0015 0.5610
## 16 0.2130 0.0016 0.0011 0.0023 0.9250
## 17 0.3550 0.0076 0.0055 0.0102 0.3480
## 18 0.8050 0.0182 0.0156 0.0204 0.3950
## 19 0.5520 0.0102 0.0092 0.0117 0.4730
## 20 0.0123 0.0008 0.0000 0.0013 0.0945
library(ggplot2)
ggplot(N[-1,], aes(x=region, y=abundance.estimate)) +
geom_bar(stat="identity", fill="#95B6C1") +
coord_flip() +
geom_errorbar(aes(ymin=abundance.lower, ymax=abundance.upper),
width=0.2, color="#105A73") +
ylab("Abundance (M males)") +
xlab("BCR") +
theme_minimal()
The densplot element contains the regional landcover specific
densities:
## pick a region
results$densplot$region
## [1] "Canada" "4" "5" "6" "7" "8" "9" "10"
## [9] "11" "12" "13" "14" "6-0" "6-1" "7-0" "7-1"
## [17] "8-0" "8-1" "8-2" "8-3"
region <- 1
results$densplot$region[region]
## [1] "Canada"
## densities for this region
(D <- as.data.frame(lapply(results$densplot$data, "[[", 1)))
## landcover estimate lower upper
## 1 Conifer 0.0066 0.0062 0.0073
## 2 Taiga Conifer 0.0032 0.0021 0.0040
## 3 Deciduous 0.0158 0.0150 0.0167
## 4 Mixedwood 0.0152 0.0146 0.0159
## 5 Shrub 0.0049 0.0043 0.0058
## 6 Grass 0.0050 0.0045 0.0053
## 7 Arctic Shrub 0.0014 0.0009 0.0020
## 8 Arctic Grass 0.0020 0.0014 0.0026
## 9 Wetland 0.0059 0.0051 0.0068
## 10 Cropland 0.0052 0.0044 0.0056
ggplot(D, aes(x=landcover, y=estimate)) +
geom_bar(stat="identity", fill="#95B6C1") +
coord_flip() +
geom_errorbar(aes(ymin=lower, ymax=upper),
width=0.2, color="#105A73") +
ylab("Density (males/ha)") +
xlab("Landcover") +
theme_minimal()
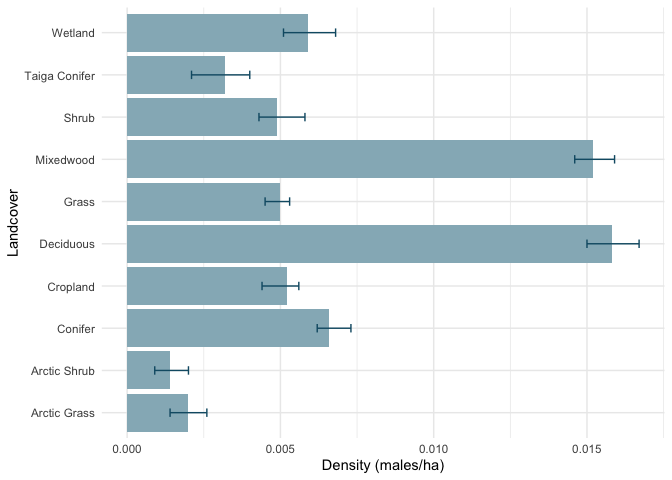
These results reflect population and density as males only (million males, males/ha, respectively). To apply a pair adjustment, the numbers have to be multiplied by 2.
Density map images
The density map images can be accessed as
https://borealbirds.github.io/api/v4/species/CAWA/images/mean-pred.png
and
https://borealbirds.github.io/api/v4/species/CAWA/images/mean-det.png.
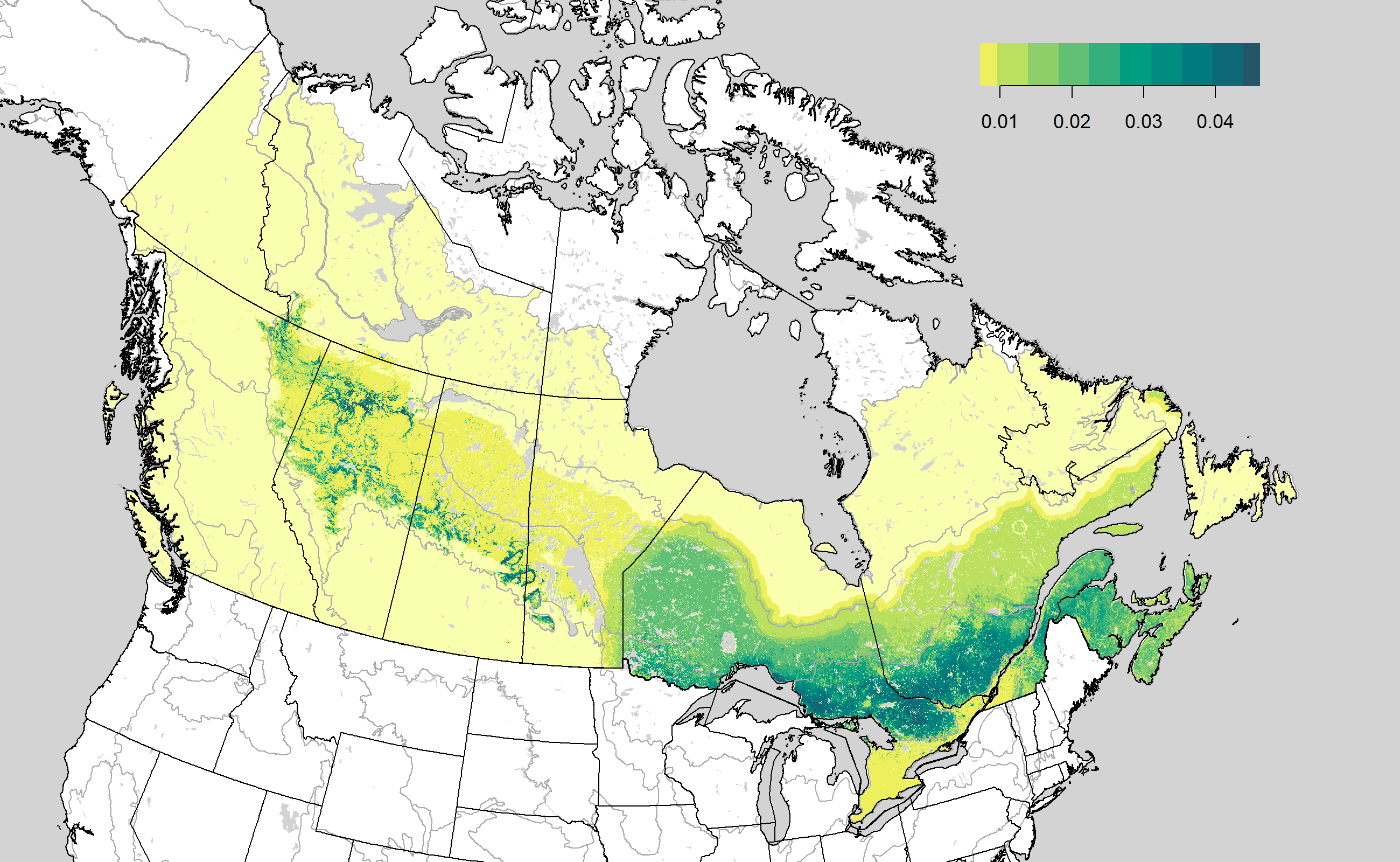
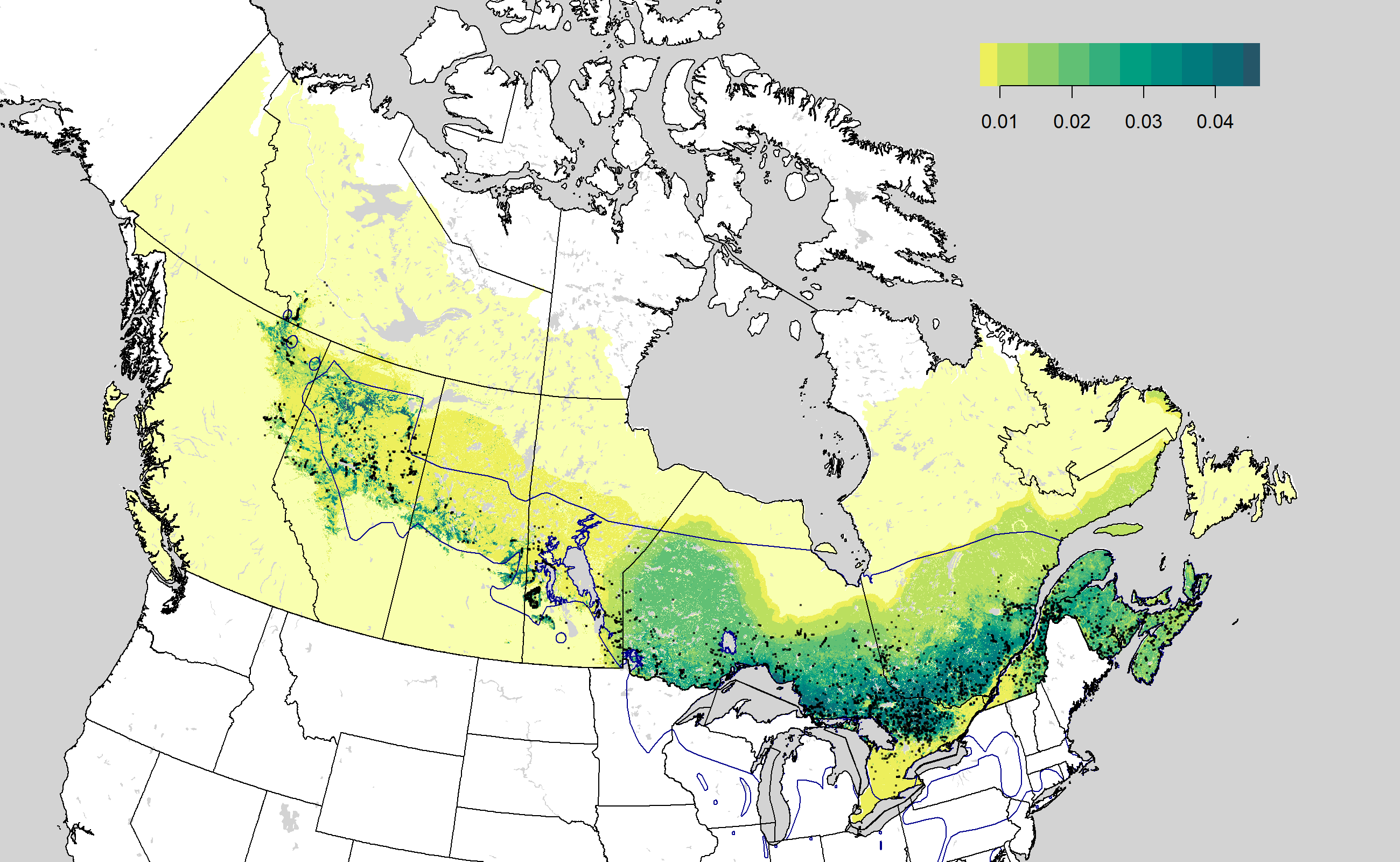
Assessing validation results
Accessing the BAMv4-results-2020-02-20.xlsx file gives us the
following tables (sheet names in parenthesis):
- metadata explaining sheets and columns (metadata)
- list species of species (species)
- list of variables (variables)
- variable importance (importance)
- model validation results (validation)
- population estimates (abundances)
- lend cover based density estimates (densities)
Download the Excel file in a temporary file:
library(readxl)
tmp <- tempfile()
download.file(file.path(api_root, "BAMv4-results-2020-02-20.xlsx"), tmp)
Now we can read in different sheets:
vars <- read_xlsx(tmp, sheet="variables")
str(vars)
## tibble [220 × 4] (S3: tbl_df/tbl/data.frame)
## $ variable : chr [1:220] "YEAR" "ARU" "AHM" "bFFP" ...
## $ definition: chr [1:220] "Year of survey" "ARU (1) or human point count (0)" "Annual heat:moisture" "Beginning of the frost free period" ...
## $ resolution: chr [1:220] NA NA "1 km" "1 km" ...
## $ source : chr [1:220] NA NA "Wang T., Hamann A., Spittlehouse D., & Carroll C. (2016) Locally Downscaled and Spatially Customizable Climate "| __truncated__ "Wang T., Hamann A., Spittlehouse D., & Carroll C. (2016) Locally Downscaled and Spatially Customizable Climate "| __truncated__ ...
Let’s read in all the tables and delete the temp file:
sheets <- c(
"metadata",
"species",
"variables",
"importance",
"validation",
"abundances",
"densities")
tabs <- list()
for (sheet in sheets)
tabs[[sheet]] <- read_xlsx(tmp, sheet)
unlink(tmp)
Here are variable importance results:
i <- tabs$importance
i <- i[i$id == spp & i$region != "Canada",]
i$BCR <- sapply(strsplit(i$region, " "), "[[", 1)
ii <- stats::xtabs(importance ~ variable + BCR, i)
heatmap(ii, margins = c(5, 10), col=hcl.colors(100, "Teal", rev=TRUE))
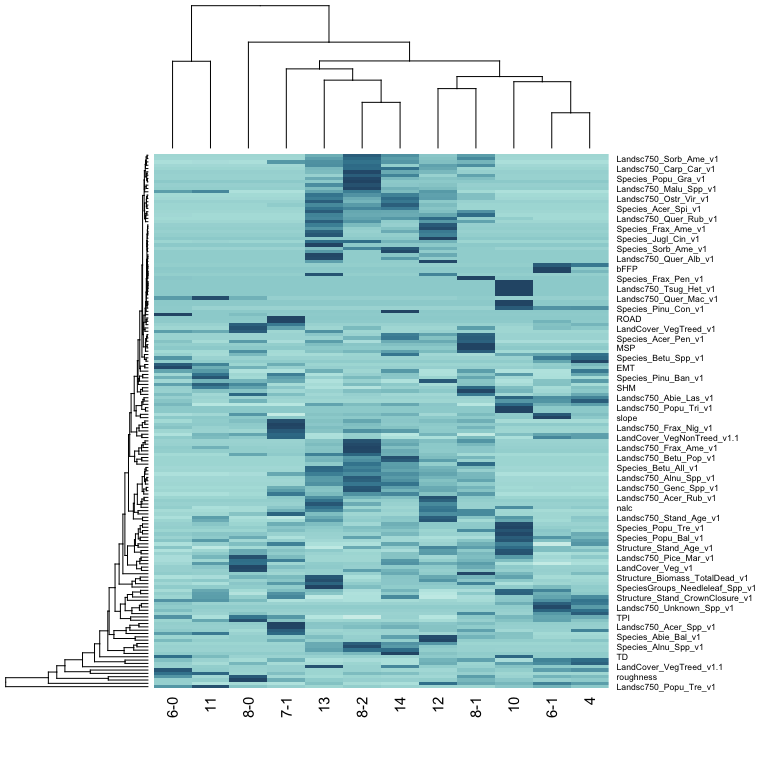
These are validation results for the Canada Warbler:
v <- tabs$validation
v <- v[v$id == spp,]
v[order(v$prevalence, decreasing=TRUE), c(4,5,8:12)]
## # A tibble: 14 x 7
## region prevalence AUC_final pseudo_R2 occc oprec oaccu
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 6-1 Boreal Taiga Plains,… 0.0215 0.934 0.265 0.596 0.664 0.898
## 2 12 Boreal Hardwood Trans… 0.0214 0.807 0.0923 0.924 0.934 0.990
## 3 8-1 Boreal Softwood Shie… 0.0204 0.770 0.0530 0.632 0.694 0.910
## 4 14 Atlantic Northern For… 0.0163 0.741 0.0640 0.826 0.862 0.958
## 5 13 Lower Great Lakes/St.… 0.0137 0.839 0.138 0.915 0.925 0.989
## 6 8-2 Boreal Softwood Shie… 0.0136 0.823 0.0879 0.598 0.666 0.897
## 7 4 Northwestern Interior … 0.0132 0.936 0.253 0.638 0.718 0.890
## 8 6-0 Boreal Taiga Plains,… 0.0122 0.857 0.165 0.861 0.890 0.967
## 9 Canada 0.0120 0.852 0.136 0.579 0.709 0.779
## 10 8-0 Boreal Softwood Shie… 0.0104 0.835 0.241 0.152 0.340 0.448
## 11 7-1 Taiga Shield & Hudso… 0.00505 0.997 -0.0723 0.0289 0.0877 0.330
## 12 11 Prairie Potholes 0.00418 0.872 0.250 0.767 0.801 0.958
## 13 10 Northern Rockies 0.00180 0.982 0.276 0.515 0.635 0.810
## 14 7-0 Taiga Shield & Hudso… 0.000426 0.0703 0 0.0754 1 0.0754
AUC was used to assess classification accuracy and pseudo R2 to quantify the proportion of variance explained. OCCC measures correspondence across bootstrap based predictions. OCCC is the product of two the overall precision (how far each observation deviated from the best fit line), and the overall accuracy (how far the best line deviates from the 1:1 line).
Working with maps
The 1 km2 resolution GeoTIFF raster files are in this shared Google Drive folder, anyone can view.
We can access the list of available files and download the files using the googledrive package:
library(googledrive)
## this should let you authenticate
drive_find(n_max = 30)
## now list files in the shared folder
f <- "https://drive.google.com/drive/folders/1exWa6vfhGo1DNUL4ei2baDz77as7jYzY?usp=sharing"
l <- drive_ls(as_id(f), recursive=TRUE)
## add species codes
l$species_id <- sapply(strsplit(l$name, "-"), "[[", 2)
## download
tmp <- tempfile(fileext = ".tif")
file_id <- l$id[l$species_id == spp]
tif_file <- drive_download(file_id, tmp)
tif_file <- drive_download(file_id,
path="~/Downloads/pred-CAWA-CAN-Mean.tif",
overwrite=TRUE)
tif_path <- tif_file$local_path
Alternatively, download the file for a species and point to its path:
tif_path <- "~/Downloads/pred-CAWA-CAN-Mean.tif"
Now we can work with the raster:
library(raster)
## Loading required package: sp
r <- raster(tif_path)
plot(r, axes=FALSE, box=FALSE, col=hcl.colors(100, "Lajolla"))
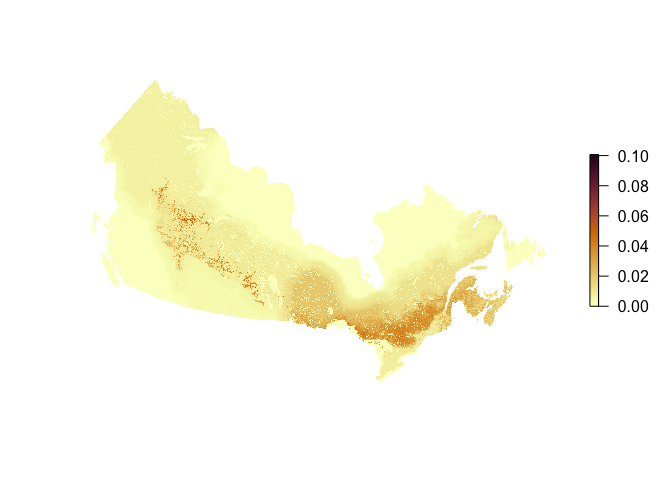
Follow this script to recreate the maps shown on the website (colors, thresholds).
Population size for custom boundary
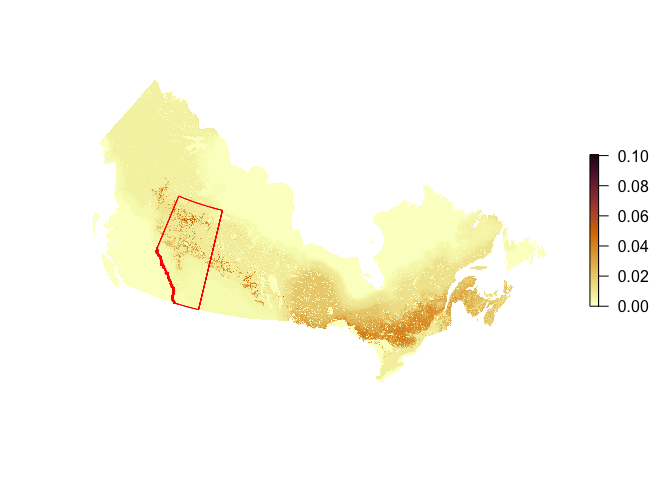
Next, we read in a custom boundary file. Let’s use the provincial boundary of Alberta now (stored as a GeoJSON file). We transform the polygon to match the projection of our raster layer and plot the two together:
library(sf)
## Linking to GEOS 3.7.2, GDAL 2.4.2, PROJ 5.2.0
#bound <- st_read("https://raw.githubusercontent.com/ABbiodiversity/cure4insect/master/inst/extdata/OSA_bound.geojson")
bound <- st_read("https://raw.githubusercontent.com/ABbiodiversity/cure4insect/master/inst/extdata/AB_bound.geojson")
## Reading layer `Alberta' from data source `https://raw.githubusercontent.com/ABbiodiversity/cure4insect/master/inst/extdata/AB_bound.geojson' using driver `GeoJSON'
## Simple feature collection with 1 feature and 1 field
## geometry type: POLYGON
## dimension: XY
## bbox: xmin: -120.0016 ymin: 48.99666 xmax: -110.0048 ymax: 60.00046
## epsg (SRID): 4326
## proj4string: +proj=longlat +datum=WGS84 +no_defs
bound <- st_transform(bound, st_crs(r))
plot(r, axes=FALSE, box=FALSE, col=hcl.colors(100, "Lajolla"))
plot(bound$geometry, add=TRUE, border="red", col=NA)
Let’s crop the density map to the extent of Alberta and mask areas outside of the boundary:
r2 <- crop(r, bound)
r2 <- mask(r2, bound)
plot(r2, axes=FALSE, box=FALSE, col=hcl.colors(100, "Lajolla"))
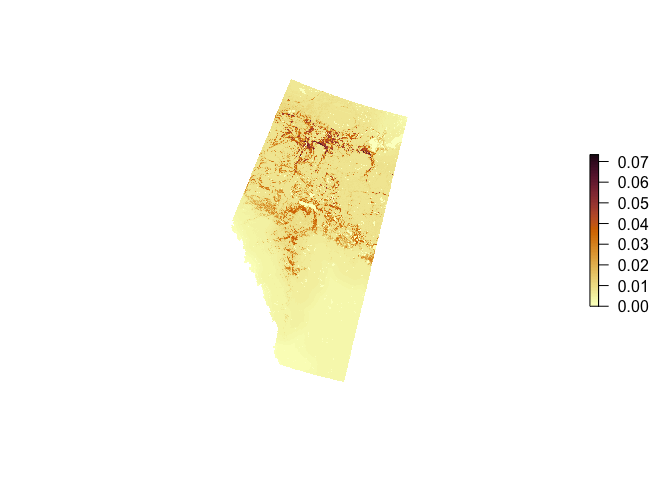
We can now sum up the raster cells to get population size (million individuals) with pair adjustment:
(N <- sum(values(r2), na.rm=TRUE) * 100 * 2) / 10^6
## [1] 1.082979
Post-stratified density estimates
Post-hoc stratification (‘post-stratification’) is an approach to estimate land cover based density estimates (males per ha) for a species based on the density map and a classification layer.
Let’s use the 2005 MODIS-based North American landcover map as an
example within the Alberta boundary. We calculate the mean of the pixel
level predicted densities (PS).
## read in raster
lc <- raster("https://raw.githubusercontent.com/ABbiodiversity/recurring/master/offset/data/lcc.tif")
## crop and mask to boundary
lc <- mask(crop(lc, bound), bound)
## extract cell values
LC <- data.frame(
lc=values(lc), # land cover classes, integer
density=values(r2), # males / ha
area=1) # km^2
## remove NA values (cells outside of boundary but inside bounding box)
LC <- LC[!is.na(LC$density),]
## land cover classes
labs <- c(
"Conifer"=1,
"Taiga Conifer"=2,
"Deciduous"=5,
"Mixedwood"=6,
"Shrub"=8,
"Grass"=10,
"Arctic Shrub"=11,
"Arctic Grass"=12,
"Wetland"=14,
"Cropland"=15)
LC$label <- names(labs)[match(LC$lc, labs)]
head(LC)
## lc density area label
## 1045 6 0.006929385 1 Mixedwood
## 1046 6 0.006855813 1 Mixedwood
## 1047 6 0.006849375 1 Mixedwood
## 1825 6 0.007014129 1 Mixedwood
## 1826 6 0.006815174 1 Mixedwood
## 1827 1 0.006703767 1 Conifer
## aggregate density by land cover
(PS <- aggregate(list(density=LC$density), list(landcover=LC$label), mean))
## landcover density
## 1 Arctic Grass 0.003469213
## 2 Conifer 0.006983323
## 3 Cropland 0.004930945
## 4 Deciduous 0.019037611
## 5 Grass 0.004553917
## 6 Mixedwood 0.013687571
## 7 Shrub 0.006586476
## 8 Taiga Conifer 0.007123922
## 9 Wetland 0.008748280
Here are the post stratified density values:
ggplot(PS, aes(x=landcover, y=density)) +
geom_bar(stat="identity", fill="#95B6C1") +
coord_flip() +
ylab("Density (males/ha)") +
xlab("Landcover") +
labs(title=paste(results$species$english, "in Alberta"),
caption="Based on post-stratification") +
theme_minimal()
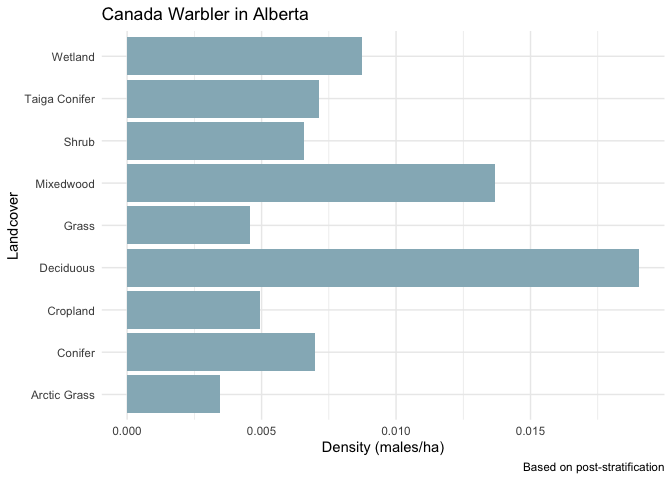
These results can be used in region specific analyses that require density values as inputs, for example landcover based scenario analyses.